

- This page was created by volunteers like you!
- Help us make it even better. To learn more about contributing to MEpedia, click here.
- Join the movement
- Visit #MEAction to find support or take action. Donate today to help us improve and expand this project.
Español:Síndromes de Ehlers-Danlos
Los Síndromes de Ehlers-Danlos (EDS, por sus siglas en inglés) son un grupo de trastornos hereditarios que afectan al tejido conectivo - principalmente la piel, articulaciones y pared de los vasos sanguíneos.[1] Son enfermedades genéticas caracterizadas por deficiencias en la producción o el metabolismo del colágeno y otras anomalías del tejido conectivo.[2][3] La severidad de la presentación varía ampliamente y se caracteriza por hiperlaxitud articular (también llamada hipermovilidad articular, que es una excesiva movilidad de las articulaciones), hiperextensibilidad cutánea (piel hiperelástica) y fragilidad tisular (es decir, de los tejidos).[4]
Signos y Síntomas
_phalangeal_joints.JPG)
Los síntomas varían ampliamente entre individuos de acuerdo al subtipo de EDS. La afectación del tejido conectivo provoca manifestaciones que van desde alteraciones leves de las articulaciones hasta aquellas que ponen en riesgo la vida del paciente.[2]
- Hiperlaxitud articular.- las articulaciones tienen un rango de movilidad mayor al normal.
- Articulaciones inestables o laxas, propensas a subluxaciones y luxaciones (dislocaciones).
- Dolor articular.
- Hiperextensibilidad articular.- las articulaciones se estiran más de lo normal.
- Artritis de presentación temprana.
- Piel suave y aterciopelada.
- Piel frágil que se desgarra o desarrolla hematomas (moretones) con facilidad.
- Cicatrices atróficas (aquella cicatriz que es de característica hundida).
- Regeneración de heridas lenta e ineficiente.
- Desarrollo de pseudotumores moluscoides.
- Dolor muscular.
- Bajo tono muscular (poco común).
Subtipos
-
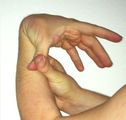 El EDS hiperlaxo (hEDS), clásico (cEDS) y semi-clásico (clEDS) se diagnostican utilizando la escala de Beighton y el criterio de tipos del EDS. En este caso se muestra una de las mediciones que corresponde a los casos en que el pulgar alcanza a tocar el antebrazo.
El EDS hiperlaxo (hEDS), clásico (cEDS) y semi-clásico (clEDS) se diagnostican utilizando la escala de Beighton y el criterio de tipos del EDS. En este caso se muestra una de las mediciones que corresponde a los casos en que el pulgar alcanza a tocar el antebrazo. -
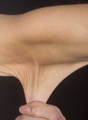 Piel hiperelástica en una persona con EDS clásico (cEDS).
Piel hiperelástica en una persona con EDS clásico (cEDS). -
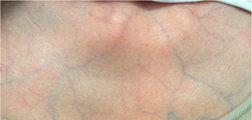 Piel translúcida de un paciente con EDS vascular (vEDS).
Piel translúcida de un paciente con EDS vascular (vEDS). -
 Cicatriz atrófica (hundida) característica de cEDS, clEDS, el tipo dermatosparaxis y otros tipos.
Cicatriz atrófica (hundida) característica de cEDS, clEDS, el tipo dermatosparaxis y otros tipos. -
 Dedos largos y delgados característicos del tipo cifoescoliótico de EDS que asemejan a los del Síndrome de Marfanoid (Marfanoid habitus, por su nomenclatura médica).
Dedos largos y delgados característicos del tipo cifoescoliótico de EDS que asemejan a los del Síndrome de Marfanoid (Marfanoid habitus, por su nomenclatura médica). -
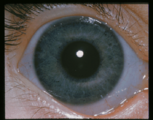 Menor de 4 años diagnosticada con Síndrome de la córnea frágil (BCS) caracterizado por párpados inflamados, protuberancia anterior de la córnea, escleras (blanco del ojo) azules, miopía alta (miopía severa) y queratocono.
Menor de 4 años diagnosticada con Síndrome de la córnea frágil (BCS) caracterizado por párpados inflamados, protuberancia anterior de la córnea, escleras (blanco del ojo) azules, miopía alta (miopía severa) y queratocono.

.png)
.png)



Los EDS se clasifican en trece subtipos que incluyen 6 diferentes tipos y subtipos claramente definidos, y 5 presentaciones clasificadas como “otros tipos”.[2][5][6]
- EDS hiperlaxo o hipermóvil (hEDS).- es la manifestación clínica más prevalente. Presenta hiperlaxitud articular (excesiva movilidad de las articulaciones) que provoca luxaciones (dislocaciones), hematomas (moretones) y dolor crónico que comúnmente es desproporcionado en comparación con los hallazgos físicos y radiológicos.
- EDS clásico (cEDS) y semi-clásico o mitis (clEDS).- se presentan con hiperextensibilidad cutánea (piel hiperelástica) pronunciada, hiperlaxitud articular (excesiva movilidad de las articulaciones) y en el caso del clEDS, facilidad en el desarrollo de hematomas (moretones).
- EDS vascular (vEDS) y EDS cardiovalvular (cvEDS).- se presentan con fragilidad arterial, intestinal o uterina con la posibilidad de ruptura del propio órgano o de la arteria correspondiente. La piel suele ser delgada o translúcida permitiendo la visualización de las venas. Los pacientes con cvEDS desarrollan problemas cardio-valvulares progresivos.
- Tipo cifoescoliótico (kEDS).- se presenta con escoliosis, laxitud articular (articulaciones laxas) e hipotonía muscular severa (bajo tono muscular severo) al nacimiento. La escoliosis es progresiva y puede resultar en la imposibilidad de caminar al alcanzar los 20 o 30 años de edad. Otras características comunes incluyen las correspondientes al síndrome de Marfanoid (Marfanoid habitus, por su nomenclatura médica), caracterizado por dedos largos y delgados; extremidades inusualmente largas; y pecho hundido o protuberante.
- Tipo artrocalásico (aEDS).- se presenta con dislocación de cadera e hiperlaxitud articular generalizada al nacimiento (congénitas). También puede presentar hiperextensibilidad cutánea (piel hiperelástica), fragilidad tisular (es decir, de los tejidos), cifoescoliosis e hipotonía muscular (bajo tono muscular).
- Tipo dermatosparaxis (dEDS).- se presenta con fragilidad cutánea severa y una gran facilidad para desarrollar hematomas (moretones).
- Otros tipos.- en esta categoría se agrupan las presentaciones genéticas menos comunes incluyendo aquellas que sólo han sido detectadas en una sola familia y son: Síndrome de la córnea frágil (BCS); Síndrome de Ehlers-Danlos espondilodisplásico EDS (spEDS); Síndrome de Ehlers-Danlos musculocontractural (mcEDS); Síndrome de Ehlers-Danlos miopático (mEDS); y Síndrome de Ehlers-Danlos periodontal (pEDS).[5][6]
Prevalencia
Los síndromes de Ehlers-Danlos (EDS) afectan tanto a hombres como a mujeres.[6]
1 de cada 5,000 personas a nivel mundial presenta alguno de los tipos o subtipos del EDS.[3] Los EDS hiperlaxo (hEDS) y clásico (cEDS) son los de mayor prevalencia. El hEDS puede llegar a afectar de 1:5,000 a 1:20,000 personas, mientras que el cEDS probablemente se presenta en 1:20,000 a 1:40,000 personas. El resto son menos frecuentes y sólo afectan a algunos pocos casos o familias a nivel mundial.[7]
Factores de Riesgo
Los síndromes de Ehlers-Danlos (EDS) son enfermedades hereditarias causadas por mutaciones genéticas en uno o más de los genes relacionados con el tejido conectivo, principalmente aquellos involucrados en el metabolismo del colágeno, una proteína importante para la estructura de músculos, piel, ligamentos, tendones, cartílago, huesos, vasos sanguíneos y otros tejidos corporales.[2][3]
Diagnóstico
El diagnóstico se logra con la exploración física que incluye una prueba de hiperlaxitud articular usando la escala de Beighton o los criterios diagnósticos de Brighton.[8][9]
Patofisiología
Los síndromes de Ehlers-Danlos (EDS) son un grupo de trastornos hereditarios del tejido conectivo caracterizados por hiperlaxitud articular (excesiva movilidad de las articulaciones), fragilidad e hiperextensibilidad cutánea (piel frágil e hiperelástica). Son el resultado de deficiencias en la producción o el metabolismo del colágeno, la principal proteína estructural de la piel y el tejido conectivo, cuyas alteraciones genéticas han sido identificadas en al menos 6 de los subtipos del EDS.[2]
Al parecer, en los tipos I y II del EDS (cEDS y clEDS) las mutaciones causantes involucran a los genes COL5A1, COL5A2 y tenascina-X (TNX), y se asume que también hay alteraciones en el gen COL1A2. A pesar de que la mitad de las mutaciones que causan cEDS y clEDS parecen afectar al gen COL5A1, un número significativo de ellas son mutaciones del tipo “sin sentido” que resultan en la degradación y por tanto baja concentración del ARNm.[2][10]
El tipo vascular (vEDS) corresponde a una mutación en el gen COL3A1 de tipo autosómica dominante (AD), es decir que sólo es necesario que uno, la madre o el padre, sea portador para poder heredar la condición a su progenie. Esta mutación resulta en la disminución en la concentración de colágeno tipo III, lo que se traduce en un incremento en la fragilidad del tejido conectivo que provoca rupturas a nivel arterial, intestinal y uterino, con la consecuente muerte prematura.[11]
El tipo cifoescoliótico (kEDS) o tipo VI se caracteriza por escoliosis, laxitud articular (articulaciones laxas) generalizada, fragilidad cutánea e hipotonía muscular severa (bajo tono muscular severo) al nacimiento. Es una mutación autosómica recesiva (AR), es decir se requiere que ambos, la madre y el padre, sean portadores para poder heredar la condición a su progenie. Se han identificado más de 20 mutaciones en el gen PLOD1 que provocan la deficiencia de la proteína LH1, esencial para el entrecruzamiento y estabilidad del colágeno tipo I/III.[2][12]
La regeneración lenta e ineficiente de heridas es una característica típica del EDS que parece derivarse de defectos a nivel de los fibroblastos. Sin embargo, las heridas pueden curarse con aplicación exógena (externa) de colágeno tipo V.[2]
Los pacientes pediátricos muestran deficiencias en tres genes de la familia de la transferasa-S del glutatión: GSTM1, GSTT1, GSTP1.[2][13]
La baja actividad de la enzima beta4GalT-7 (beta4-galactosiltransferasa-7) se asocia a una presentación del EDS parecida a la progeria de Hutchinson-Gilford que provoca que las y los niños envejezcan de manera acelerada.[2]
Las mutaciones bialélicas (en ambos alelos) del gen FKBP14 pueden resultar en una presentación recesiva del EDS con cifoescoliosis progresiva, miopatía, pérdida del oído y posiblemente un aumento en el riesgo de complicaciones vasculares.[2][4]
En el sitio web de la organización Ehlers Danlos Society puede encontrar una tabla que incluye el patrón hereditario AD/AR (autosómico dominante/ autosómico recesivo), las bases genéticas y las proteínas alteradas en cada uno de los subtipos de EDS.[6]
Afectaciones asociadas al EDS
Los órganos y sistemas afectados, y trastornos asociados con el EDS, incluyen:
- Alteraciones articulares y musculares.
- Alteraciones de la piel.
- Alteraciones del Cerebro y la columna vertebral.
- Deficiencia de las válvulas del corazón.
- Diverticulitis.
- Hemorragias.
- Hernias.
- Problemas de la vesícula biliar.
- Problemas dentales, orales y de voz.
- Problemas oculares.
- Problemas del sistema cardiovascular y circulatorio.
- Subluxaciones y luxaciones (dislocaciones).
- Trastorno de ansiedad y depresión.
- Trastornos digestivos.
- Trastornos reproductivos. [14][15][16][17][18][19][20][21][22][23][24][25][26]
Comorbilidades y complicaciones
Encefalomielitis Miálgica/Síndrome de Fatiga Crónica (EM/SFC o ME/CFS por sus siglas en inglés)
En una serie de casos de adolescentes que fueron referidos a la clínica de Encefalomielitis Miálgica/ Síndrome de Fatiga Crónica (EM/SFC) del Dr Peter Rowe en 1999 se encontraron 12 pacientes que cumplían con los criterios diagnósticos del EDS y presentaban intolerancia ortostática (taquicardia postural ortostática o hipotensión ortostática neurogénica). Esto le llevó a la conclusión de que “un subgrupo de pacientes con EM/SFC e intolerancia ortostática también presenta EDS”.[27] El Dr. Rowe también describió hiperlaxitud articular (escala de Beighton >4) en 60% de los pacientes pediátricos con EM/SFC comparado con el 24% de los pacientes control.[28] En un estudio posterior en 2022, el Dr. Rowe reportó que la hiperlaxitud articular no mostró asociación alguna con otras características clínicas de la EM/SFC, a pesar de ser un factor de riesgo para su desarrollo.[29]
Un estudio sueco reportó que de los 234 pacientes diagnosticados con EM/SFC de acuerdo con los Criterios del Consenso de Canadá, el 49% presenta hiperlaxitud articular, mientras que el 20% cumple con los criterios de hEDS.[30]
Otras comorbilidades y complicaciones
- Cifosis y Escoliosis.[31]
- Disautonomía.
- Distonía. [31]
- Hipertensión intracraneal. [31]
- Inestabilidad craneocervical. [31]
- Inestabilidad atlantoaxial (subluxación atlantoaxial). [31]
- Malformaciones de Chiari. [31]
- Prolapso de la válvula mitral.
- Síndrome de activación mastocitaria (MCAS, por sus siglas en inglés).
- Síndrome de médula anclada. [31]
- Síndrome de taquicardia postural ortostática (POTS, por sus siglas en inglés).[32][33][34]
- Siringomielia. [31]
- Quistes de Tarlov (quistes extradurales). [31]
Tratamiento
Terapia farmacológica
A la fecha no existe una cura para el EDS y los tratamientos están limitados a analgésicos que no requieren receta médica como el paracetamol/acetaminofén (Tylenol®, Panadol®, FeverAll® y Tempra®), ibuprofeno (Advil®, Motrin®, Brufen®, Dalsy®, Ibudol®, Nurofen®, Algidrin®, Apirofeno®, Dolencar®, Dolorac®) y naproxeno sódico (Aleve®, Anaprox®, Anaprox DS®, Naprelan®, Naprosyn®, Antalgin®, Lundiran®, Momen®, Flanax®). Los medicamentos más potentes que requieren receta médica se utilizan en caso de heridas agudas y dolor crónico. En algunas ocasiones se prescriben medicamentos que disminuyen la presión arterial en un intento de liberar el estrés de los vasos sanguíneos.[35]
Fisioterapia (kinesiología)
El tratamiento principal para evitar las luxaciones (dislocaciones) del EDS se basa en ejercicios que ayudan a fortalecer los músculos y estabilizar las articulaciones. Los aparatos ortopédicos ayudan a prevenir luxaciones articulares (dislocaciones articulares).[35] Sin embargo, en los pacientes con EM/SFC es necesario diagnosticar la presencia y severidad de la exacerbación de síntomas post-esfuerzo (PEM, por sus siglas en inglés) y sólo en su ausencia proceder con una terapia física que debe ser gradual e individualizada.
Intervenciones quirúrgicas y otros procedimientos
Es posible recomendar cirugía en caso de luxación (dislocación) de articulaciones pero se corre el riesgo de que el tejido conectivo no sane adecuadamente. La cirugía también puede ser necesaria en caso de ruptura de órganos o vasos sanguíneos en pacientes con vEDS.[35]
Véase también
- Colágeno
- Joint hypermobility and hypermobility syndromes|Hiperlaxitud articular y síndromes de hiperlaxitud/hipermovilidad
Más información
- Herramientas para manejar el Síndrome de Ehlers-Danlos.[36]
- Embarazo, parto y alimentación para pacientes con Síndrome de Ehlers-Danlos o del espectro de trastornos de hiperlaxitud/hipermovilidad.[37]
- 2016, Una Pieza Más del Rompecabezas: un paciente de EM/SFC/Fibromialgia obtiene diagnóstico de Síndrome de Ehlers-Danlos.[38]
- Vasos Sanguíneos - Estructura de la Pared de Venas y Arterias.
References
- ↑ "Ehlers-Danlos syndrome - Symptoms and causes". Mayo Clinic. Retrieved August 17, 2018.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 "What are the Ehlers-Danlos Syndromes?". The Ehlers Danlos Society. Retrieved October 7, 2018.
- ↑ 3.0 3.1 3.2 Mao, Jau-Ren; Bristow, James (May 1, 2001). "The Ehlers-Danlos syndrome: on beyond collagens". Journal of Clinical Investigation. 107 (9): 1063–1069. ISSN 0021-9738. PMID 11342567.
- ↑ 4.0 4.1 "What are the Ehlers-Danlos Syndromes?". The Ehlers Danlos Society. Retrieved October 7, 2018.
- ↑ 5.0 5.1 "Ehlers-Danlos syndromes". Genetic and Rare Diseases Information Center. Retrieved October 6, 2018.
- ↑ 6.0 6.1 6.2 6.3 "The Types of EDS". The Ehlers Danlos Society. Retrieved August 17, 2018.
- ↑ "Ehlers-Danlos syndrome". Genetics Home Reference. Retrieved October 7, 2018.
- ↑ "Assessing Joint Hypermobility". The Ehlers Danlos Society. Retrieved August 17, 2018.
- ↑ "The Brighton Criteria for JHS". Hypermobility Syndromes Association. Retrieved August 17, 2018.
- ↑ Schwarze, U; Atkinson, M; Hoffman, GG; Greenspan, DS; Byers, PH. "Null alleles of the COL5A1 gene of type V collagen are a cause of the classical forms of Ehlers-Danlos syndrome (types I and II)". reference.medscape.com. Medscape. Retrieved October 6, 2018.
- ↑ Eder, J; Laccone, F; Rohrbach, M; Guinta, C; Aumayr, K; Reichel, C; Trautinger, F. "A new COL3A1 mutation in Ehlers-Danlos syndrome type IV". reference.medscape.com. Medscape. Retrieved October 6, 2018.
- ↑ Yeowell, HN; Walker, LC. "Mutations in the lysyl hydroxylase 1 gene that result in enzyme deficiency and the clinical phenotype of Ehlers-Danlos syndrome type VI". reference.medscape.com. Medscape. ISSN 1096-7192. Retrieved October 6, 2018.
- ↑ Kuz'mina, NS; Shipaeva, EV; Semyachkina, AN; Vasil'eva, IM; Kovalenko, LP; Durnev, LP; Zasukhina, GD. "Polymorphism of detoxification genes and cell resistance to mutagens in patients with Ehlers-Danlos syndrome". reference.medscape.com. Medscape. ISSN 0007-4888. Retrieved October 6, 2018.
- ↑ "Mental health – The Ehlers-Danlos Support UK". ehlers-danlos.org. Retrieved October 6, 2018.
- ↑ "Brain and spine – The Ehlers-Danlos Support UK". ehlers-danlos.org. Retrieved October 7, 2018.
- ↑ "Dental, oral and voice problems – The Ehlers-Danlos Support UK". ehlers-danlos.org. Retrieved October 7, 2018.
- ↑ "Digestive disorders – The Ehlers-Danlos Support UK". ehlers-danlos.org. Retrieved October 7, 2018.
- ↑ "Ehlers-Danlos Syndrome Commonly Associated Health Problems | EDSAwareness.com". chronicpainpartners.com. Retrieved October 6, 2018.
- ↑ "Joint problems – The Ehlers-Danlos Support UK". ehlers-danlos.org. Retrieved October 7, 2018.
- ↑ "Ocular Complications of Ehlers Danlos Syndrome". Total Eye Care. Retrieved October 6, 2018.
- ↑ "Ehlers-Danlos Syndrome - Children's Health Issues". MSD Manual Consumer Version. Retrieved October 7, 2018.
- ↑ "Postural tachycardia syndrome (PoTS)". The Ehlers-Danlos Support UK. Retrieved October 7, 2018.
- ↑ "Erectile Dysfunction (ED): Causes, Treatment, and More". Healthline. Retrieved October 7, 2018.
- ↑ Hurst, B.S.; Lang, M.B.; Kullstam, S.M.; Usadi, R.S.; Matthews, M.L.; Marshburn, P.B. (2012). "Reproductive challenges in women with Ehlers-Danlos syndrome: survey results from over 1350 respondents from the Ehlers-Danlos National Foundation". Fertility and Sterility. 98 (3): S112. doi:10.1016/j.fertnstert.2012.07.411. ISSN 0015-0282.
- ↑ "Skin". The Ehlers-Danlos Support UK. Retrieved October 7, 2018.
- ↑ "Dislocation/Subluxation Management". The Ehlers Danlos Society. Retrieved October 7, 2018.
- ↑ Rowe, Peter C.; Barron, Diana F.; Calkins, Hugh; Maumenee, Irene H.; Tong, Patrick Y.; Geraghty, Michael T. (October 1, 1999). "Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome". The Journal of Pediatrics. 135 (4): 494–499. doi:10.1016/S0022-3476(99)70173-3. ISSN 0022-3476.
- ↑ Barron, Diana F.; Cohen, Bernard A.; Geraghty, Michael T.; Violand, Rick; Rowe, Peter C. (September 1, 2002). "Joint hypermobility is more common in children with chronic fatigue syndrome than in healthy controls". The Journal of Pediatrics. 141 (3): 421–425. doi:10.1067/mpd.2002.127496. ISSN 0022-3476.
- ↑ Vogel, Sarah K.; Primavera, Isabelle R.; Marden, Colleen L.; Jasion, Samantha E.; Cranston, Erica M.; Flaherty, Marissa A.K.; Violand, Richard L.; Rowe, Peter C. (January 2022). "The Presentation of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome Is Not Influenced by the Presence or Absence of Joint Hypermobility". The Journal of Pediatrics. 240: 186–191.e2. doi:10.1016/j.jpeds.2021.09.014. ISSN 1097-6833. PMID 34537220.
- ↑ "Bragee Bertilson et al. - ME CFS and Intracranial Hypertension". Center for Open Science. November 27, 2019. Retrieved December 3, 2019.
- ↑ 31.0 31.1 31.2 31.3 31.4 31.5 31.6 31.7 31.8 Henderson, Fraser C.; Austin, Claudiu; Benzel, Edward; Bolognese, Paolo; Ellenbogen, Richard; Francomano, Clair A.; Ireton, Candace; Klinge, Petra; Koby, Myles (February 21, 2017). "Neurological and spinal manifestations of the Ehlers-Danlos syndromes". American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 175 (1): 195–211. doi:10.1002/ajmg.c.31549. ISSN 1552-4868.
- ↑ "The Ehlers-Danlos Society is forming a Comorbid Condition Coalition! - Inspire". inspire.com. November 8, 2017. Retrieved October 7, 2018.
- ↑ "What is Craniocervical Instability? - The Pain Relief Foundation". The Pain Relief Foundation. Retrieved October 7, 2018.
- ↑ "Neurological and Spinal Manifestations of the Ehlers-Danlos Syndromes (for Non-experts) | The Ehlers Danlos Society". The Ehlers Danlos Society. Retrieved October 7, 2018.
- ↑ 35.0 35.1 35.2 "Ehlers-Danlos syndrome - Diagnosis and treatment - Mayo Clinic". Mayo Clinic. Retrieved October 7, 2018.
- ↑ "Ehlers Danlos Syndromes Toolkit". rcgp.org.uk. Royal College of General Practitioners. Retrieved October 29, 2018.
- ↑ "Pregnancy, birth, feeding and hypermobile Ehlers-Danlos syndrome / hypermobility spectrum disorders – The Ehlers-Danlos Support UK". ehlers-danlos.org. Retrieved October 7, 2018.
- ↑ Burns, Darden (July 1, 2016). "Another Piece of the Puzzle: An ME/CFS/FM Patient Gets an Ehlers Danlos Syndrome Diagnosis - Health Rising". Health Rising. Retrieved August 17, 2018.